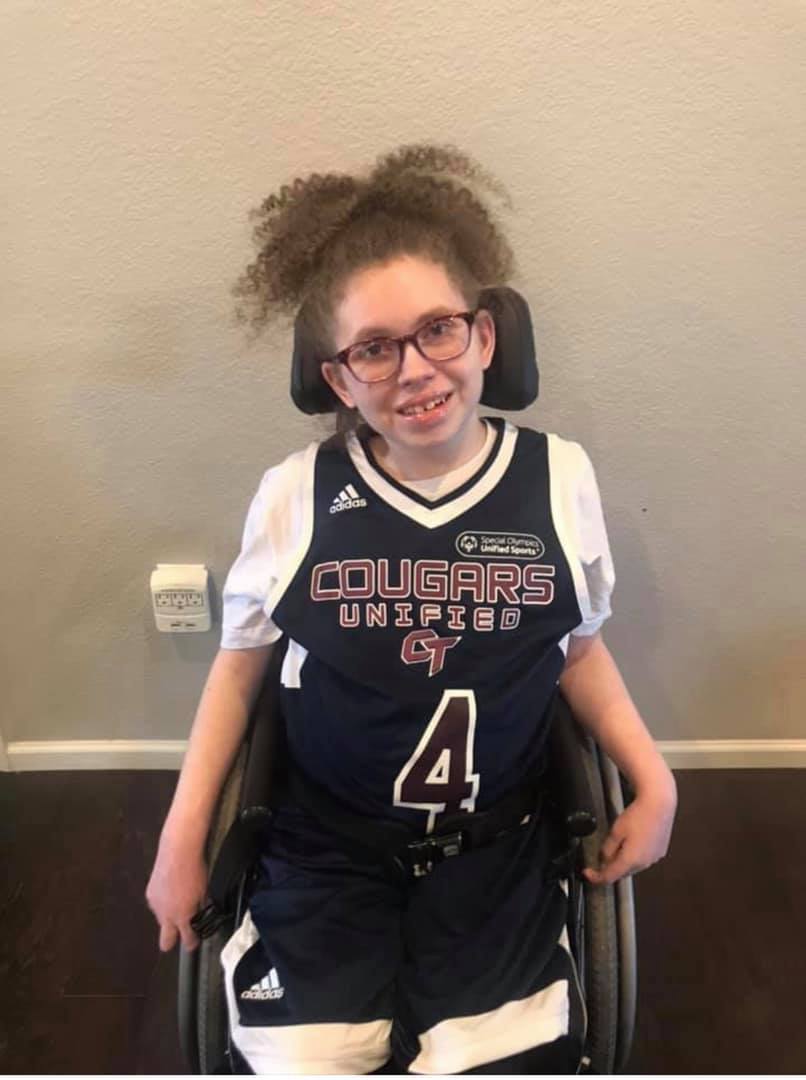
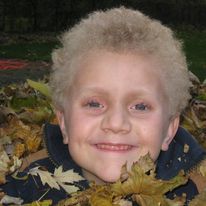
GIANT AXONAL NEUROPATHY
What is GAN?
Giant Axonal Neuropathy (GAN) is a recessively inherited condition that results in progressive nerve death. GAN is a loss of function disorder, meaning an intracellular protein needed for long-term nerve survival either isn’t being made or is being immediately degraded before it can perform its job. Every gene in the body has a function. Some genes encode or make secreted enzymes. Some genes make intracellular proteins. The protein that isn’t functioning in GAN is called gigaxonin. It is believed that the role of gigaxonin is to degrade its binding partners when they are done doing their job on the axon portion of the nerve. In the absence of functional gigaxonin, these other proteins are toxically communicating, causing densely packed, swollen giant axons, which interfere with the signaling between the peripheral nervous system (arms, legs, etc) and the central nervous system (brain and spinal cord). It’s easiest to think of a clogged vacuum hose.
Giant axonal neuropathy generally appears in early childhood. It progresses slowly as neuronal injury becomes more severe. Signs of giant axonal neuropathy first appear in the peripheral nervous system. These symptoms most often include clumsiness, lack of coordination (impaired proprioception), low muscle tone and impaired fine motor skills are caused by nerve damage in the central, autonomic and peripheral nervous systems. The motor neurons and sensory neurons are impacted by GAN and these nerves govern movement and sensation in the arms, legs, and other parts of the body. The autonomic nervous system controls involuntary processes like heart rate, body temperature, and communication with organs such as the lungs for involuntary breathing. Most individuals with this disorder first have problems with walking. Later they may lose sensation, coordination, strength, and reflexes in their limbs. Hearing and visual problems typically occur. Extremely kinky hair (as compared to others in the family) is characteristic of giant axonal neuropathy, but not all confirmed GAN cases have kinky hair.
As the disorder progresses, patients become quadriplegic, dependent on a feeding tube and ventilator before dying typically in the second or third decade.
How Common is Giant Axonal Neuropathy
Giant axonal neuropathy is a very rare disorder; the incidence is unknown. We now have a few milder progressing, confirmed GAN cases, as well as a few confirmed cases without kinky hair. MDs working with HHF now feel that some CMT Type 2 patients who have not yet had their causal gene identified may actually have GAN. If you have been clinically diagnosed with CMT and told you have nystagmus (involuntary eye movements when looking sideways) and/or optic motor atrophy and wish to have your GAN gene sequenced for free, please contact HHF to arrange for sequencing.
")
Known GAN Cases by Location
How do People Inherit Giant Axonal Neuropathy?
GAN is a recessive genetic disorder. Each gene in the body has a role. Sadly, the GAN gene is life-sustaining. It is required for long-term nerve survival.
At conception, we inherit one copy of each gene from each parent. When parents are carriers of a mutation on the same gene, there’s a 25% chance they will have an offspring with the disorder. Commonly, to have a child with GAN, both parents are carriers of a mutation of one copy of their GAN gene – an autosomal recessive pattern.
There are a few GAN cases resulting from uniparental disomy, meaning that one parent passed two copies of their defective GAN gene to their offspring. At conception, if an extra copy of a gene is passed, there’s quality control that will kick out one copy, sometimes resulting in uniparental disomy. Therefore, it is possible to have an offspring with a recessive genetic disorder with only one parent being a carrier.
What Genes are Related to Giant Axonal Neuropathy?
Mutations in a single gene called the Giant Axonal Neuropathy (GAN) gene.
Giant axonal neuropathy is caused by mutations in the GAN gene, which provides instructions for making a protein called gigaxonin. Some GAN mutations change the shape of the protein, affecting how it binds to other proteins to form a functional complex. Other mutations prevent cells from producing any gigaxonin protein.
Gigaxonin is involved in a cellular function that destroys and gets rid of excess or damaged proteins using a mechanism called the ubiquitin-proteasome system. Neurons without functional gigaxonin accumulate excess neurofilaments in the axon, causing the axons to become distended. These giant axons do not transmit signals properly and eventually deteriorate, resulting in problems with movement and other nervous system dysfunction.
READ MORE
HELP US FIND A TREATMENT AND CURE
If there are any GAN patients out there who are nearing the end of their fight, please consider having an autopsy performed by a neuropathologist. Please make certain Hannah’s Hope Fund gets a copy of the autopsy report. Through the gift of one autopsy a few years ago, we’ve learned a great deal more about the impact of GAN on the central nervous system. Since then, our team of scientists have discovered this disease has an early profound impact on sensory and autonomic nerves in the peripheral nervous system. Please, please do this for the other children who still have a chance for treatment and cure. Hannah’s Hope Fund will pay for the autopsy if there’s a fee.
Please donate generously to help us find treatment and a cure.
”Together we can achieve anything”
USEFUL GAN LINKS
Scroll through to see the many faces of GAN